

Expectation
- Text-guided Protein Design
Fengyuan Dai, Yuliang Fan, Jin Su, Chentong Wang, Chenchen Han, Xibin Zhou, Jianming Liu, Hui Qian, Shunzhi Wang, Anping Zeng, et al. Toward de novo protein design from natural language. bioRxiv, pages 2024–08, 2024.
Shengchao Liu, Yutao Zhu, Jiarui Lu, Zhao Xu, Weili Nie, Anthony Gitter, Chaowei Xiao, Jian Tang, Hongyu Guo, and Anima Anandkumar. A text-guided protein design framework. arXiv preprint arXiv:2302.04611, 2023.
- Advanced Protein ChatGPT
Chao Wang, Hehe Fan, Ruijie Quan, and Yi Yang. Protchatgpt: Towards understanding proteins with large language models. arXiv preprint arXiv:2402.09649, 2024.
Method
蛋白质-功能对的构建
对于给定的蛋白质,我们利用 UniProt 数据库 [7] 中几乎所有子条目的描述信息来构建蛋白质-功能对。我们将蛋白质的子条目信息分为两类:序列水平 和 残基水平(见补充表 2)。
- 序列水平信息:由一条或多条完整句子组成,描述蛋白质的整体特征,如其功能和催化活性。
- 残基水平信息:包含关于蛋白质序列中特定残基的描述性片段,如结合位点和活性位点,这些信息不能直接使用。
对于残基水平的子条目,我们使用 GPT-4 [39] 生成多个特定模板句子,从而将零散的信息组织成连贯的句子。对于序列水平的子条目,我们同样利用 GPT-4 对描述进行 释义,并生成多个备选句子,以增强模型对文本输入的鲁棒性。最终,一个蛋白质的每个子条目都会生成一条文本句子,并与该蛋白质配对,形成 蛋白质-功能对,用于训练或评估。
预训练数据集的构建
首先,我们在经过人工审核的 Swiss-Prot 数据库 [5] 上执行了 50% 序列相似性聚类。其中,选择 1000 个簇作为验证集,另 1000 个簇作为测试集,其余数据用作训练集。对于每个蛋白质,我们按照上述方法构建蛋白质-功能对,最终得到 1400 万条高质量的蛋白质-功能对,并以此训练了 ProTrek 的初始版本。该初始模型为 35M 参数规模,在 12 张 NVIDIA 80G A100 GPU 上运行 100K 步完成预训练。
接着,我们利用初始 ProTrek 模型对 UniRef50 数据库 [7] 中的 3 亿条蛋白质-功能对进行打分与筛选,保留所有得分高于 Swiss-Prot 平均分的蛋白质-功能对,最终得到 2500 万条数据。将这 2500 万条与原始的 1400 万条合并,构成了 最终的预训练数据集。
预训练损失函数
我们采用 InfoNCE 损失函数 [43] 来实现蛋白质序列–结构–功能的对比学习。InfoNCE 损失函数可以表示为:
$L_{InfoNCE}=-log\frac{exp(f(z_i,z_j)/\tau)}{\sum_{k=1}^Nexp(f(z_i,z_k)/\tau)}$
其中,$z_i$和 $z_j$ 表示任意两种模态的嵌入,$f(z_i,z_j)$ 表示嵌入之间的相似性分数,$N$ 为批次中的配对总数,$\tau$ 为可学习的温度参数。我们分别针对 序列–结构、序列–功能、结构–功能 三种模态配对,从两个方向计算 InfoNCE 损失,共得到 6 个对比学习损失函数。
此外,我们还在 ProTrek 的 序列编码器 和 结构编码器 中引入了 掩码语言模型(Masked Language Modeling, MLM) [40] 的损失函数,以保持模型在氨基酸和 3Di token 水平上的识别能力。其训练目标是通过捕捉被掩码位置与周围上下文之间的依赖关系来预测被掩码的 token,损失函数形式化表达为:
$L_{MLM}=\sum_{i\in T}-logP(s_i|S_{\setminus T})$
其中,T 表示被掩码的 token 位置集合,$S_{\setminus T}$表示在特定位置 token 被掩码的蛋白质/结构序列。
最终的损失函数由 2 个 MLM 损失函数 与 6 个 InfoNCE 损失函数 的平均值构成。
预训练设置
我们将 ProTrek 的 序列编码器 初始化为 ESM-2 650M [19],文本编码器 初始化为 PubMedBERT [15],而 结构编码器 因无可用的预训练模型,则采用随机初始化。
在优化过程中,我们使用 DeepSpeed 策略 [44] 并采用 AdamW 优化器 [42],其超参数设置为:
- $\beta_1$=0.9,$\beta_2$=0.98;
- L2 权重衰减设为 0.01。
学习率在前 2000 步 内从 0 逐步升至 4e-4,随后通过 余弦退火调度 [41] 降至 4e-5。整个训练过程共持续约 100K 步,在 20 张 NVIDIA 80G A100 GPU 上完成。
在输入处理上,我们将蛋白质序列与结构序列 截断至最多 512 个 token,而测试描述文本则 截断至最多 100 个 token。整体批量大小为 1280 个蛋白质-序列-结构-功能配对。此外,训练过程中我们采用了 混合精度训练 来加速模型训练。
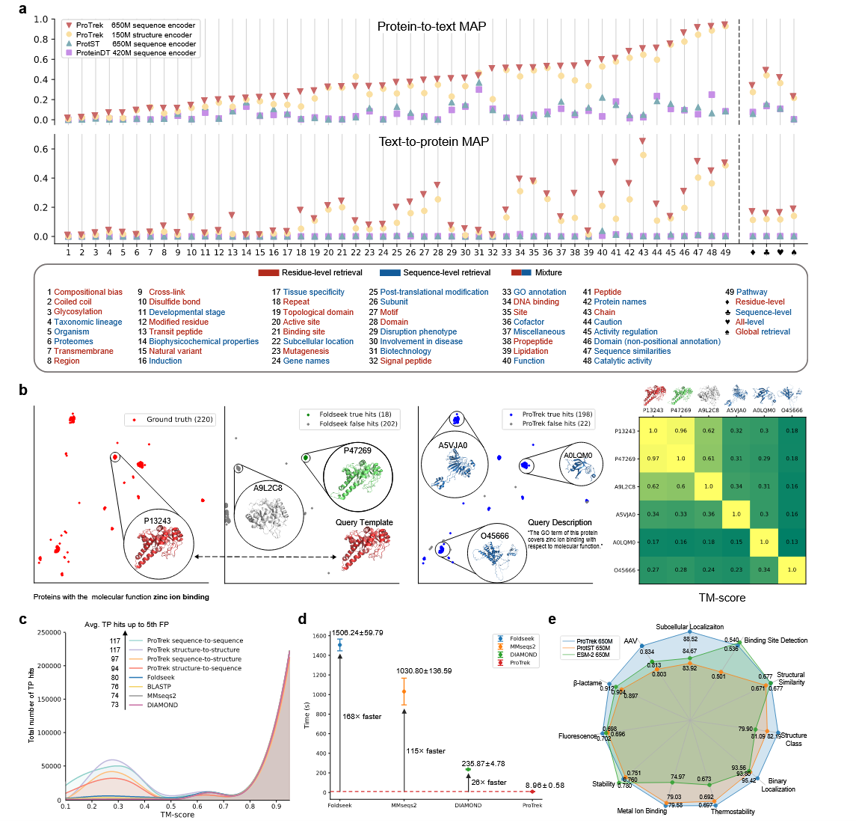
蛋白质-功能检索基准
我们利用 Swiss-Prot 测试集中的 4,000 个蛋白质构建了蛋白质-功能检索基准。为评估 ProTrek 在其他数据库蛋白质上的泛化性能,我们在测试集中额外加入了 从 UniProt 随机采样的 100,000 个蛋白质作为负样本。所有这些蛋白质的文本描述被加入文本集合中。
在 蛋白质到文本检索任务中,模型需要检索与 Swiss-Prot 测试集蛋白质最匹配的功能描述。相反,在 文本到蛋白质检索任务中,模型需要在接近 104,000 个负样本蛋白质的背景下,从 Swiss-Prot 中找到与查询功能描述(仅使用人工审阅的描述作为查询文本)匹配的蛋白质。
为减少配对数量较少的子部分对评估偏差的影响,我们选择了 配对数量最多的前 33 个功能子部分进行评估。
检测功能相似的蛋白质
在 ProTrek 及所有基线方法中,我们基于同一 Swiss-Prot 测试集(4,000 个蛋白质)进行了 全对全搜索(all-versus-all search),并比较了它们在寻找 具有相同 GO 注释的蛋白质时的性能。
对于给定的查询蛋白质,其 GO 注释集合记为 $G_q$。同样地,对于数据库中检索到的每个候选结果,其 GO 注释集合记为 $G_h$。若存在至少一个共同的 GO 注释,即满足:$G_q \cap G_h \neq \emptyset$
则认为该结果是 正确命中(correct hit)。
在图 2c 中,y 轴的数值由 所有查询蛋白质的正确命中数汇总得到。
Mean Average Precision(MAP)
平均平均精度(Mean Average Precision, MAP) 是信息检索任务中常用的指标,其定义为所有查询的 平均精度(AP) 的平均值。
对于给定查询,AP 的计算公式为:
$AP = \frac{1}{R} \sum_{i=1}^{N} P(i) \cdot r(i)$
其中:
- $R$表示相关结果的数量;
- $N$ 表示结果总数;
- $P(i)$ 表示排名列表中第$i$个位置的精度;
- $r(i)$表示第$ i $个结果是否相关(相关为 1,不相关为 0)。
最终,MAP 定义为所有查询的 AP 的平均值:
$AP = \frac{1}{Q} \sum_{q=1}^{Q} AP_q$
深度学习基线模型
在蛋白质-功能检索任务中,我们将 ProTrek 与 ProtST [35] 中的 ProtST-ESM-1b 以及 ProteinDT [20] 中的 ProtBERT BFD-512-1e-5-1e-1-text-512-1e-5-1e-1-InfoNCE-0.1-batch-9-gpu-8-epoch-5 进行比较。
在下游蛋白质任务中,我们使用 ESM-2 650M 版本 [19] 和 ProtST-ESM-1b 作为基线模型。
下游任务微调
我们严格遵循 SaprotHub [27] 的微调设置,包括数据集预处理、划分及微调超参数等。我们对所有模型参数进行微调直至收敛,并根据其在验证集上的表现选择最佳检查点。
工具运行命令
Foldseek
我们使用了 Foldseek 的版本 ef4e960ab84fc502665eb7b84573dfff9c2aa89d。命令行在默认参数下执行:
1 | foldseek easy-search pdb dir targetDB aln.m8 tmpFolder |
MMseqs2
我们使用了 MMseqs2 的版本 edb8223d1ea07385ffe63d4f103af0eb12b2058e。命令行在默认参数下执行:
1 | mmseqs easy-search seqs.fasta targetDB alnRes.m8 tmp |
BLASTP
我们使用了 Protein-Protein BLAST 2.15.0+ 版本。命令行运行方式如下:
1 | blastp -query seqs.fasta -db db -outfmt 6 -out blastp_result |
DIAMOND
我们使用了 DIAMOND v2.1.9.163 版本。在 very-sensitive 模式下运行,命令行如下:
1 | diamond blastp -q seqs.fasta -d db -o result.tsv --very-sensitive -k 0 |

